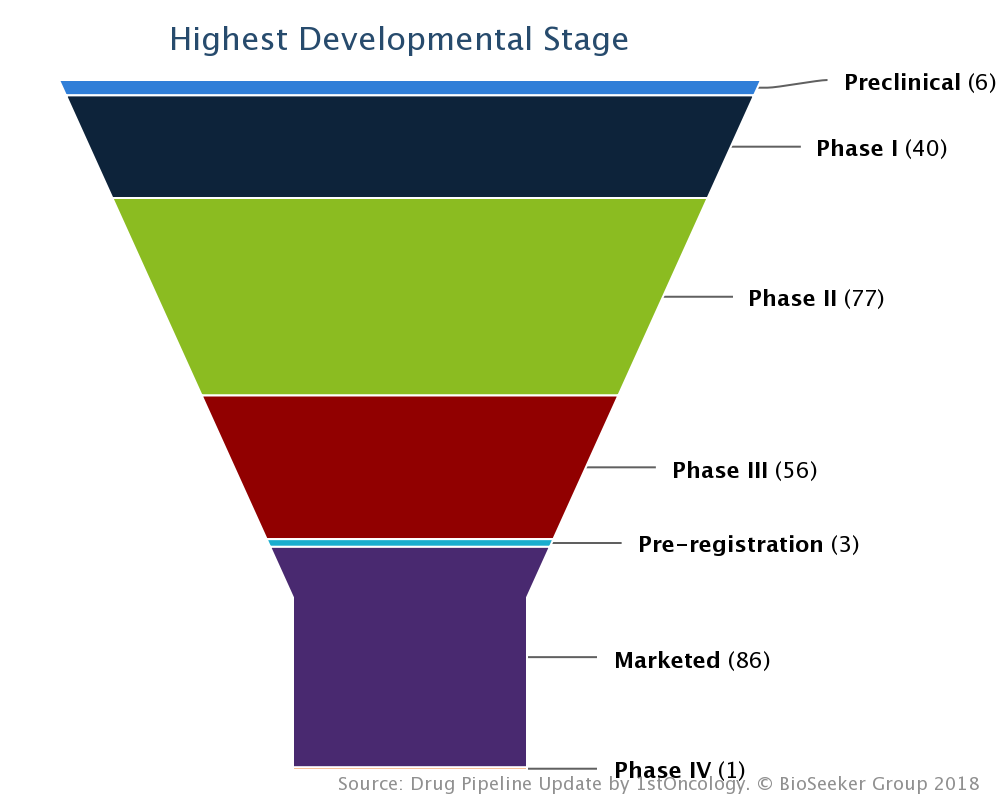
On October 19, 2018 – Novartis reported presentation of a new analysis of Lutathera (lutetium Lu 177 dotatate*) NETTER-1 data at the 2018 European Society for Medical Oncology (ESMO) (Free ESMO Whitepaper) congress examining the impact of Lutathera treatment on patients with low, medium or high liver tumor burden (Press release, Novartis, OCT 19, 2018, View Source [SID1234529967]). The data show that Lutathera treatment results in significant improvement in progression free survival (PFS) regardless of the extent of baseline liver tumor burden (LTB), elevated alkaline phosphatase (ALP) liver enzyme or presence of large (>30mm diameter) lesion in patients with progressive midgut neuroendocrine tumors (NETs) compared to octreotide LAR alone[1].
Schedule your 30 min Free 1stOncology Demo!
Discover why more than 1,500 members use 1stOncology™ to excel in:
Early/Late Stage Pipeline Development - Target Scouting - Clinical Biomarkers - Indication Selection & Expansion - BD&L Contacts - Conference Reports - Combinatorial Drug Settings - Companion Diagnostics - Drug Repositioning - First-in-class Analysis - Competitive Analysis - Deals & Licensing
Schedule Your 30 min Free Demo!
Lutathera is the first Peptide Receptor Radionuclide Therapy (PRRT) to receive regulatory registration, with approval by the European Commission in September 2017 and by the US Food and Drug Administration (FDA) in January 2018. Lutathera is an Advanced Accelerator Applications product.
"Patients with metastatic midgut NET and a high liver tumor load at diagnosis have a poorer prognosis than patients with few liver metastases[2],[3]," said Jonathan Strosberg, MD, Associate Professor, Section Head, Neuroendocrine Tumor Program at Moffitt Cancer Center, and Principle Investigator of the NETTER-1 study. "These new data provide hope for these patients and reinforce the potential benefit of Lutathera treatment in this population."
Liver tumor burden (LTB) was defined as tumor volume/total liver volume by CT or MRI, and categorized as low (<25%), moderate (25-50%), and high (>50%)[1]. Median PFS (months) in Lutathera arm vs 60 mg octreotide LAR alone was 28.35 vs 11.04 in low (HR=0.218, 95% CI 0.120 to 0.394); Not Reached (NR) vs 8.67 in moderate (HR=0.202, 95% CI 0.077 to 0.525); 19.38 vs 5.52 in high LTB (HR= 0.193, 95% CI 0.079 to 0.474), respectively[1].
Because the numbers of patients and events of deteriorations are small for the moderate and high liver burden groups for quality of life assessments, moderate/high liver burden groups were pooled into one group[1]. Median TTD (months) for global health status (self-assessment of overall health and quality of life) in Lutathera arm vs 60 mg octreotide LAR alone was 28.81 vs 6.11 in low (HR=0.376, 95% CI 0.196 to 0.720); and NR vs 5.98 in moderate/high LTB (HR=0.453, 95% CI 0.178 to 1.152) [1]. Median TTD (months) for physical functioning was 25.20 vs 11.47 in low (HR=0.512, 95% CI 0.264 to 0.994); and NR vs 11.56 in moderate/high LTB (HR=0.526, 95% CI 0.207 to 1.335) [1].
"These results from the NETTER-1 study continue to show that Lutathera delivers strong efficacy in patients with a challenging disease burden," said Samit Hirawat, MD, Head of Novartis Oncology Global Drug Development. "Demonstrating improved PFS and maintenance of QoL in patients with neuroendocrine tumors with poor prognosis due to a high liver tumor burden is a great example of our commitment to reimagining cancer."
Additional sub-analysis evaluated median PFS in patient subgroups with normal or elevated baseline levels of liver enzyme alkaline phosphatase (ALP), and in subgroups with presence or absence of a large (>30 mm diameter) lesion at baseline[1]. Median PFS (months) in Lutathera arm vs 60 mg octreotide LAR alone for the group with normal ALP was 28.35 vs 8.74 (HR=0.204, 95% CI 0.117 to 0.357)[1]. Median PFS (months) in Lutathera arm vs 60 mg octreotide LAR alone for the group with elevated ALP was NR vs 5.78 (HR=0.191, 95% CI 0.090 to 0.405)[1]. Median PFS (months) in Lutathera arm vs 60 mg octreotide LAR alone for the group with a large tumor lesion was 28.35 vs 8.44 (HR=0.266, 95% CI 0.165 to 0.429)[1]. Median PFS (months) in Lutathera arm vs 60 mg octreotide LAR alone for the group without a large tumor lesion was NR vs 8.74 (HR= 0.069, 95% CI 0.021 to 0.233)[1].
The NETTER-1 trial is an international phase III study in patients with progressive, somatostatin receptor-positive midgut neuroendocrine tumors[4]. Patients were randomized to treatment with Lutathera (Lu)(n=117) and best supportive care (30 mg octreotide LAR), or 60 mg octreotide LAR alone (Oct) (n=114). In total, 141 patients had low (71 Lu, 70 Oct), 50 patients had moderate (19 Lu, 31 Oct), and 40 patients had high LTB (27 Lu, 13 Oct).
European Organisation for Research and Treatment of Cancer (EORTC) quality of life questionnaires, a commonly used metric for analysis of HRQoL in cancer patients, were assessed during the trial to determine the impact of treatment on HRQoL[5]. Patients completed the questionnaires at baseline and every 12 weeks until tumor progression. TTD was defined as the time from randomization to the first QoL deterioration >=10 points (on a 100-point scale) compared to baseline score for the same domain.
About Lutathera
Lutathera (lutetium Lu 177 dotatate*) is a lutetium Lu 177-labeled somatostatin analog peptide. Lutathera belongs to a class of treatments called Peptide Receptor Radionuclide Therapy (PRRT). Lutathera is comprised of a targeting molecule which carries a radioactive component. Lutathera has received orphan drug designation from the FDA and the European Medicines Agency (EMA). In the US, Lutathera is indicated for the treatment of somatostatin receptor positive gastroenteropancreatic neuroendocrine tumors (GEP-NETs), including foregut, midgut, and hindgut neuroendocrine tumors in adults[6]. In Europe, Lutathera is indicated for the treatment of unresectable or metastatic, progressive, well differentiated (G1 and G2), somatostatin receptor positive gastroenteropancreatic neuroendocrine tumors (GEP-NETs) in adults[7]. Lutathera can cause serious side effects that may include bone marrow problems, kidney problems, liver problems, hormonal gland problems, fertility problems and problems arising from radiation exposure. Please see Important Safety Information and Full Prescribing Information at: www.lutathera.com.
* USAN: lutetium Lu 177 dotatate / INN: lutetium (177Lu) oxodotreotide